As an RNA virus, New Coronavirus is considered to be prone to mutation during transmission. Scientists have tracked through the past two months that, as of now, there are 13 accumulating mutations in the S protein (spike glycoprotein) of the new coronavirus, and one of the mutant virus strains is occupying a dominant advantage. The S protein is the “key” for the new coronavirus to enter human cells, and it is also the target of most vaccine strategies and antibody-based treatments.
The above findings are from the well-known Los Alamos National Laboratory (Los Alamos National Laboratory), Duke Institute of Human Vaccine and Surgery, University of Sheffield The team’s researchers published a study published on the preprint platform bioRxiv on May 5 local time: “Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2.”
The study ’s corresponding author and lead author is Bette Korber, a computational biologist at Los Alamos National Laboratory. It has made significant contributions to obtaining effective HIV vaccines, one of which is currently undergoing human trials in Africa. In 2004, Korber won the Ernest Orlando Lawrence Award (Ernest Orlando Lawrence Award), which is the highest recognition of scientific achievements awarded by the US Department of Energy.

The research team developed an analysis pipeline to track mutations in SARS-CoV-2 in real time. They believe that considering mutations in a broader phylogenetic context, geographic location, and time can provide an “early warning system” to reveal the selective advantages that mutations may bring in the spread or resistance to interventions.
The paper wrote that one of the mutations the research team tracedResearch on the mutation of the S protein D614G of the virus strain is imminent. It began to spread in Europe in early February. When it appears in a new area, it will soon become the main spreading virus. This mutant virus, which originated in Europe or an important promoter of the global pandemic. In addition, the research team confirmed that recombination occurs between the strains, suggesting multiple strains of infection. These findings are of great significance to the spread, pathogenesis and immune intervention of SARS-CoV-2.
However, there are also views expressing doubts about the above conclusions, that this research has not been peer-reviewed and needs to be treated with caution. The challenger suggested that it is too early to judge whether any strain is more contagious. Some people even suggested that the reason why the G614 (late research) mutant virus spread so far and wide is because it happens to infect areas where no measures were taken during the initial stage of the pandemic. Perhaps researchers need to do more work to determine whether any strain is more infectious than other strains, and to determine whether the rate of virus mutation may exceed the speed of vaccine development.
“There are a lot of speculations here, and they have not been experimentally verified.” said Dr. Peter Hotez of the Vaccine Development Center at Texas Children’s Hospital in Houston.
Dr. Bill Hanage of Harvard University ’s Chen Zengxi School of Public Health believes that single point mutations are unlikely to have any effect. Hanage wrote that it is important to remember that any drugs or vaccines must be tested to fight any virus that is spreading. “The virus can find escape mutations on all these vaccines so early, which is too lucky.” He also believes that because few people have any natural immunity to the virus, there is little or no selection pressure that causes escape mutations. .
Of course, the research team led by Korber emphasized in the paper that in view of the importance of S protein in virus infectivity and as an antibody target, it is urgent to pass the “Early Early Warning “to assess the evolution of S protein. The main purpose of this study was to identify the dynamic patterns of mutations. In addition, based on recombination is an important aspect of coronavirus evolution, the research team also tried to determine whether recombination played a role in the evolution of SARS-CoV-2 outbreaks.
There is an urgent need to evaluate S protein evolution through “early warning”
There is an urgent need to evaluate S protein evolution through “early warning”
The paper mentioned that although the currently observed SARS-CoV-2 sequence is diverse Sex is low, but its rapid spread as a virusProvide sufficient opportunities for natural selection to produce fewer mutations that are beneficial to themselves. This is similar to the situation with influenza, where the virus slowly accumulates mutations on hemagglutinin protein (HA) throughout the influenza season. “SARS-CoV-2 is new to us, we don’t yet know if it will seasonally weaken as the weather warms and humidity increases, but we lack immunity and it has a higher spread compared to the flu Ability, these may be the reasons why it will not weaken. “
A bad situation that must be considered is that if the outbreak does not subside, the first vaccine In the previous year or longer, antigenic drift and the accumulation of immune-related mutations in the population will worsen the epidemic. The so-called antigen drift refers to that the virus changes its gene to change the sequence and structure of the protein, so that people who are originally immune cannot recognize it.
The research team believes that this concern is reasonable, and now focusing on this risk may be able to seize the important evolutionary change of the virus, “If you ignore this change, you may end up It makes the effectiveness of the first batch of vaccines in clinical use discounted. “
At present, most of the global vaccines against SARS-CoV-2 target the S protein, so as to obtain protection Sexual neutralizing antibody. S protein mediates the combination and invasion of virus and host cells. It consists of S1 domain and S2 domain, which mediate receptor binding and membrane fusion, respectively. Because the duration of the outbreak is not long enough to conduct the effectiveness test of the SARS-CoV vaccine, we currently lack key information that will help guide the development of the SARS-CoV-2 vaccine.
In the past two months, the Los Alamos National Laboratory HIV Database team turned to the new crown study, they will “Global Shared Influenza Data Initiative” (GISAID) SARS-CoV-2 sequence data in the database as a benchmark, developed an analysis pipeline (analysis pipeline) to track the evolution of SARS-CoV-2S protein in real time. The analysis software (www.cov.lanl.gov) was developed by the Los Alamos National Laboratory in cooperation with the Duke University Neutralizing Antibody Evaluation Group.
It is worth noting that the research team observed that there are actually fewer mutations between SARS-CoV-2 sequences, which limits the traditional sequence-based method To test the applicability of positive selection. However, they propose that another analysis framework can be used to determine the positive choice, which is based on GISAID database, GISAID provides a rich database of thousands of sequences linked to geographic information and sampling dates. This makes it possible to track the early signs of positive selection by identifying changes in mutation frequency that change over time. In this study, the research team analyzed 4535 (as of April 13) complete S protein sequences in the GISAID database.
Since S protein mutations are still rare, the research team has set a low threshold for loci, which is regarded as an “interest locus” for further tracking. Therefore, when a mutation was found in 0.3% of the sequence, the research team began to track it by studying its evolutionary trajectory and modeling its structural effects. For example, it has Potential impact of glycosylation pattern.
class = “contheight”>
According to the above low threshold criteria, the research team tracked a total of 13 sites and a local mutation cluster . The paper discusses a particularly interesting mutation D614G, and then briefly summarizes other tracking sites. They initially noticed that “the frequency of D614G is increasing at an alarming rate, which shows that it has an adaptive advantage over the original strain and can spread faster.”
G614 mutant virus strain has become the main epidemic strain worldwide
G614 mutant virus strain has become the main epidemic strain worldwide
mutation D614G (change in GA base at 23403 in Wuhan reference strain) The team only tracked the site mutation in the first S protein mutation report in early March; it was found 7 times out of the 183 sequences available at the time.
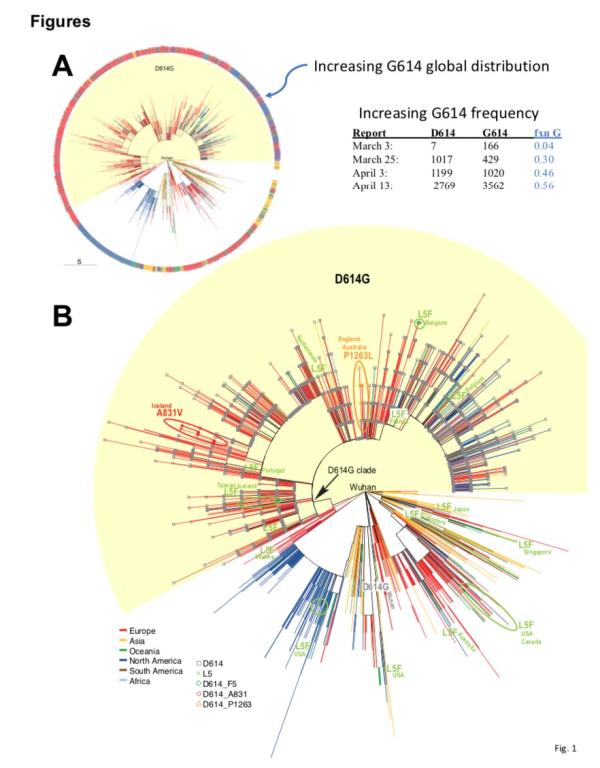
class = “contheight “>
4 of these 7 first batches of D614G mutant virus strains were sampled in Europe, and in Mexico, Brazil and Wuhan One sample was taken from each of them. Among the 5 strains, the D614G mutation was also accompanied by two other mutations: CT mutation at position 3037 of the nsp3 gene, and CT mutation at position 14409, resulting in an amino acid change in RNA-dependent RNA polymerase (RdRp) (RdRp P323L). The paper points out that the combination of these three mutations formed the basis of the branch that will soon appear in Europe. The earliest European mutation D614G appeared in Germany (EPI_ISL_406862), the sampling time was January 28, the virus strain There are also CT mutations at 3037 and no mutations at 14409.
In the research team ’s second mutation report in mid-March, due to the high frequency of occurrence in GISAID , D614G continues to be tracked and is called the “G” branch. It is worth noting that this virus is found in 29% of the samples in the world, but it is almost only found in Europe.
The data available for research in mid-March shows that between sampling and reporting The two-week lag time is consistent with the “ancestor effect” that may occur in Europe. The entire European continent subsequently spreads and the number of European samples in the GISAID database also increases.
< / div> However, data from GISAID in early April showed that the frequency of the G614 mutant increased at an alarming rate throughout March, and it is clearly seen that its geographic distribution continues to expand.

They observed that a clear and consistent pattern was observed in almost every place where there was enough sampling. That is, when the COVID-19 epidemic began in most countries and states, that is, the sampling sequence before March 1st showed that the D614 mutant was dominant. However, once G614 appears in a place, it will broadcast quickly in the crowd, and in most cases it will become the dominant virus strain in the area within a few weeks.
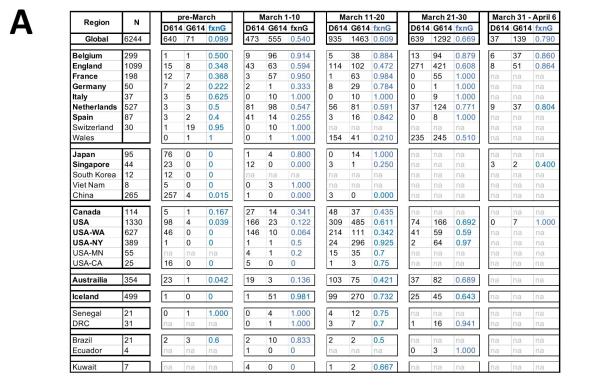
In Europe where G614 initially began to spread widely, D614 and G614 spread together early in the outbreak, but except Italy and Switzerland, D614 is more common in most sampling countries . Throughout March, G614 became more common in Europe and became dominant in April.
In North America, the initial infection was mainly in the form of D614, but in early March, G614 appeared in Canada and the United States, and became the two countries by the end of March. The main form of virus. Washington State is the state with the most SARS-CoV-2 sequences available in the GISAID database in the United States. It exemplifies the above pattern. Over time, many other states have also seen similar changes. Sampling in New York is relatively scarce. Sequence uploading began in March, and G614 is already the main form. At that time, the form of G614 also became prominent in other parts of the United States.
Regarding the situation in Europe and North America, the research team believes that it is unclear whether the local pandemic was “sown” by European contacts, or whether it has gained high Popular American contacts “sow seeds”, or a combination of these two paths.
Australia, like the United States and Canada, has transitioned from D614 to G614.
Iceland is the only exception. Iceland has undergone extensive testing. The epidemic appears to start in the form of G614, but there is a short-term increase in D614, but then it continues to be constant Low level.
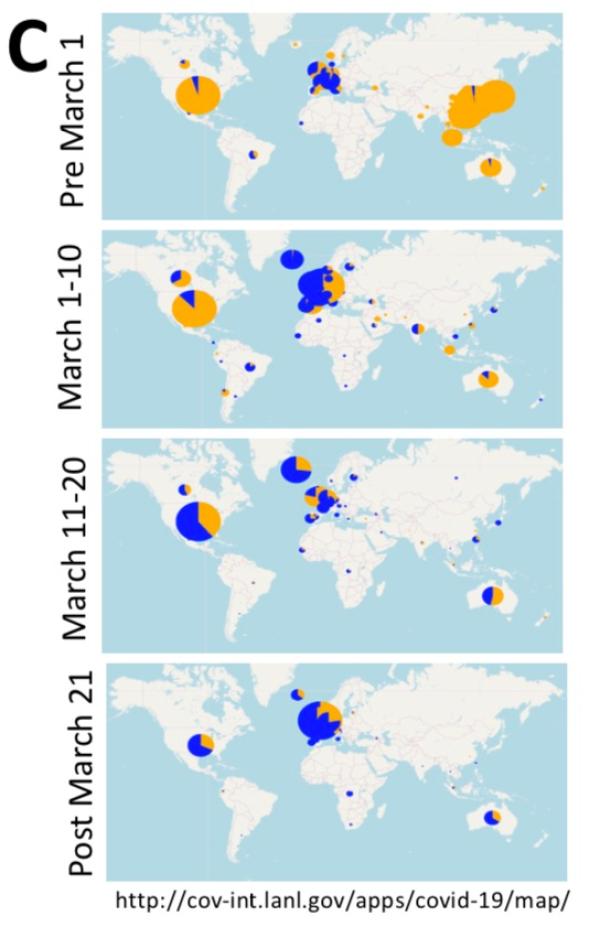
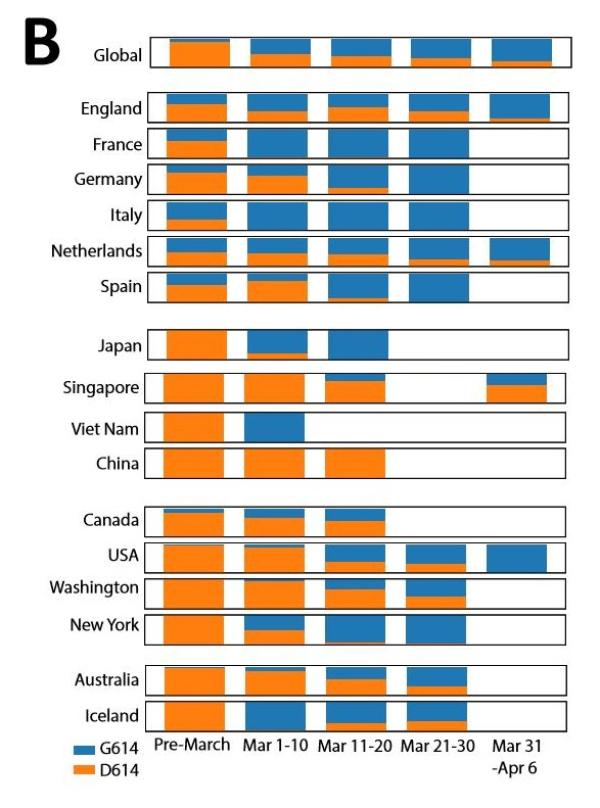
Asian samples are completely dominated by the D614 form originally detected by Wuhan, but by mid-March, G614 form was clearly established and spread in Asian countries outside of China. In addition, it should be noted that since after March 1, as the epidemic gradually subsided in China, few sequences from China were uploaded to GISAID, and the status of China’s D614G mutation is still unclear.
In addition, there are still very few samples from South America and Africa.
In summary, the research team believes that the data shows that G614 has a selective advantage, and all the pandemics in various regions occur within a few weeks. D614 dominates to G614 dominates The change reflects this.
The research team also mentioned that the G614 branch virus was also found four times in early samples in China. A sequence from Wuhan (EPI_ISL_412982) sampled on February 7 had the D614G mutation, but no other two mutations occurred at the same time. They speculated that this single D614G mutation may have occurred independently.
The remaining three cases may be related to the German sequence. A sequence sampled on January 24 was from Zhejiang (EPI_ISL_422425), which had all three mutations related to the G branch that spread in Europe. The other two samples were from Shanghai, and were sampled on January 28 and February 6 (EPI_ISL_416327, EPI_ISL_416334). As with the German sequence, they did not show a mutation at 14409.
The research team believes that considering that these early Chinese sequences are highly correlated with German sequences in the entire genome, the current global pandemic G614 may have originated in China , But it may have originated in Europe because it appeared in both regions at the end of January.
In addition, there is no recent GISAID sequence from Shanghai or Zhejiang, so the study is notIt is clear whether G614 has a selective advantage in Shanghai, but D614 became popular in Shanghai in January and early February.
Patients carrying the G614 mutation have a higher viral load, but it does not significantly aggravate the disease
Patients carrying the G614 mutation have a higher viral load, but it does not significantly aggravate the disease
What is the underlying mechanism behind the enhanced adaptability of D614G? The research team proposed that there are two different conceptual frameworks that can explain why the D614G mutation is associated with increased transmission.
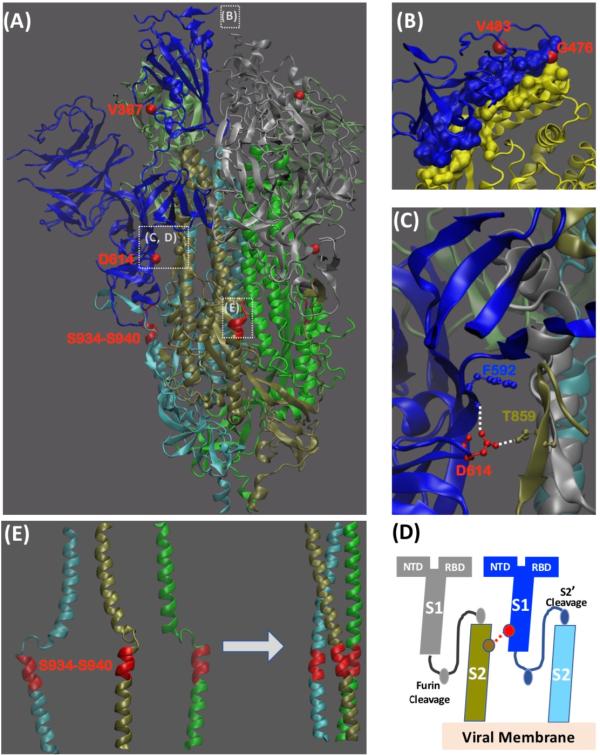
The first one is based on structure. D614 is located on the surface of the S protein propolymer and can be in contact with the adjacent propolymer. The cryo-electron microscopy structure indicates that the side chain of D614 may form hydrogen bonds with the adjacent propolymer T859. This propolymer-propolymer hydrogen bond may be crucial, as it can combine the residues of the S1 domain of one propolymer with the S2 domain of another propolymer. The D614G mutation reduced the interaction between S1 and S2, and promoted the separation of S1 and S2. This mutation may also affect the binding of RBD-ACE2. However, the research team emphasized that more detailed experimental and modeling studies are needed to clarify the effect of this mutation on RBD transformation.
The second way D614G mutations may affect transmission is immunity. D614 is embedded in the immunodominant linear epitope of the original SARS-CoV S protein. The peptide has a high serological response (64%) and induced a long-term b-cell memory response in the convalescent serum of infected individuals during the 2002 SARS-CoV epidemic. Whether in vitro or in vivo, antibodies against this peptide mediate the antibody-dependent enhancement (ADE) of SARS-CoV infection through an epitope sequence-dependent mechanism. The so-called ADE refers to the phenomenon that the virus can significantly increase the ability of replication or infection with the help of corresponding antibodies. Studies have speculated that antibody binding may mediate the conformational change of the S protein, thereby increasing the interaction of RBD-ACE2 and producing an enhancement effect.
Therefore, based on existing informationThe research team believes that the D614G mutation may affect the infectivity of S in several ways: it may increase receptor binding, fusion activation, or ADE antibody induction. Another mechanism may mediate antibody escape only through antigen drift.
It is worth noting that the sensitivity of the modified S protein to early neutralizing antibodies may also be reduced, which may make the susceptible people suffer a second infection.
The research team also considered another question. If the D614G mutation can increase the ability to spread, can it also affect the severity of the disease? Because clinical outcome data is not available in GISAID, the research team focused on a single geographic area, Sheffield, England. Sheffield followed the patterns observed in most parts of the world, starting with D614 at the beginning and turning to G614 by the end of March. They studied 453 patients with COVID-19 from Sheffield.
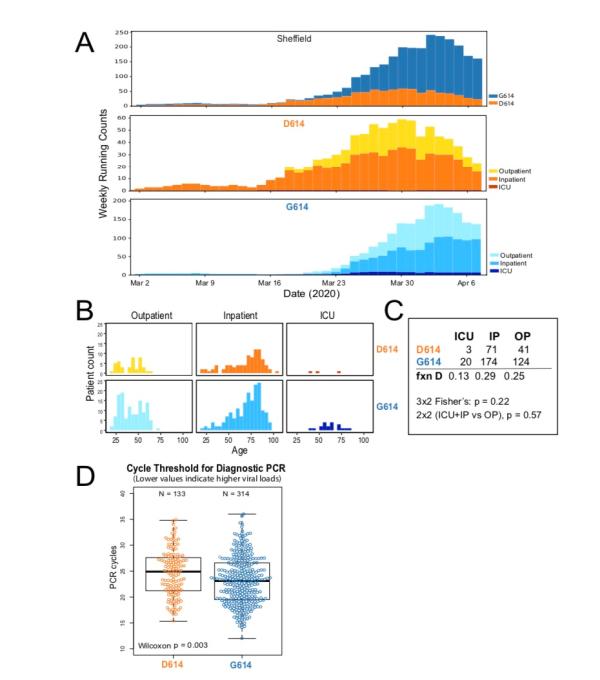
The results show that patients with the G614 mutation have a higher viral load compared to D614. But this does not necessarily mean an increased risk of disease. They mentioned that although the G614 mutation is slightly enriched in ICU patients, it is not statistically significant.