Bats are natural reservoirs for various coronaviruses (CoVs) such as SARS-Cov, SARS-Cov-2, etc. However, the scientific community has evolved and Little is known about diversity. The research team used the Bayesian statistical framework and sequence data of all known bat coronaviruses in China (including 630 new coronavirus gene sequences) to analyze their macro-evolution, cross-species transmission, and distribution across China.
The research team found that compared with the β-coronavirus, the host of the α-coronavirus switches more frequently, and can cross other hosts with more distant relationships, while the β- Coronavirus is more restricted by the host. The results showed that host switching between the families and between the genera of Rhinolophus and Rhinolophidae was the most common. This analysis also identified the host taxonomic group and geographic area of the evolutionary diversity hotspots of Chinese coronaviruses.
The research team believes that this research may help to discover bat coronavirus for active surveillance of zoonotic diseases. They suggested that SARS-CoV-2 may have originated from the genus Lycoperthus, and compared with the Lycoperthus bat, full-length genomic analysis showed that the Malay pangolin is unlikely to be the origin of the new crown virus.
The corresponding author of the study is Professor Peter Daszak, a disease ecologist of the American non-profit organization Environmental Ecology and Health Alliance, and director of the Center for Emerging Infectious Diseases, Wuhan Institute of Virology, Chinese Academy of Sciences Researcher Shi Zhengli, Director of the Key Laboratory of Highly Pathogenic Pathogen Biology and Biosafety, Chinese Academy of Sciences.
The study included 630 partial sequences of the coronavirus RNA-dependent RNA polymerase (RdRp) gene with a size of about 440bp. These sequencing samples were from various ChineseBat anal swabs or feces collected locally; in addition, it also includes 608 sequences from the Chinese bat coronavirus gene sequence published in GenBank by different research teams.
The research team used the Coronavirus RdRp gene sequence data group to reconstruct the phylogenetic tree by Bayesian method and analyzed the ancestors of bat alpha-coronavirus and beta-coronavirus Host.
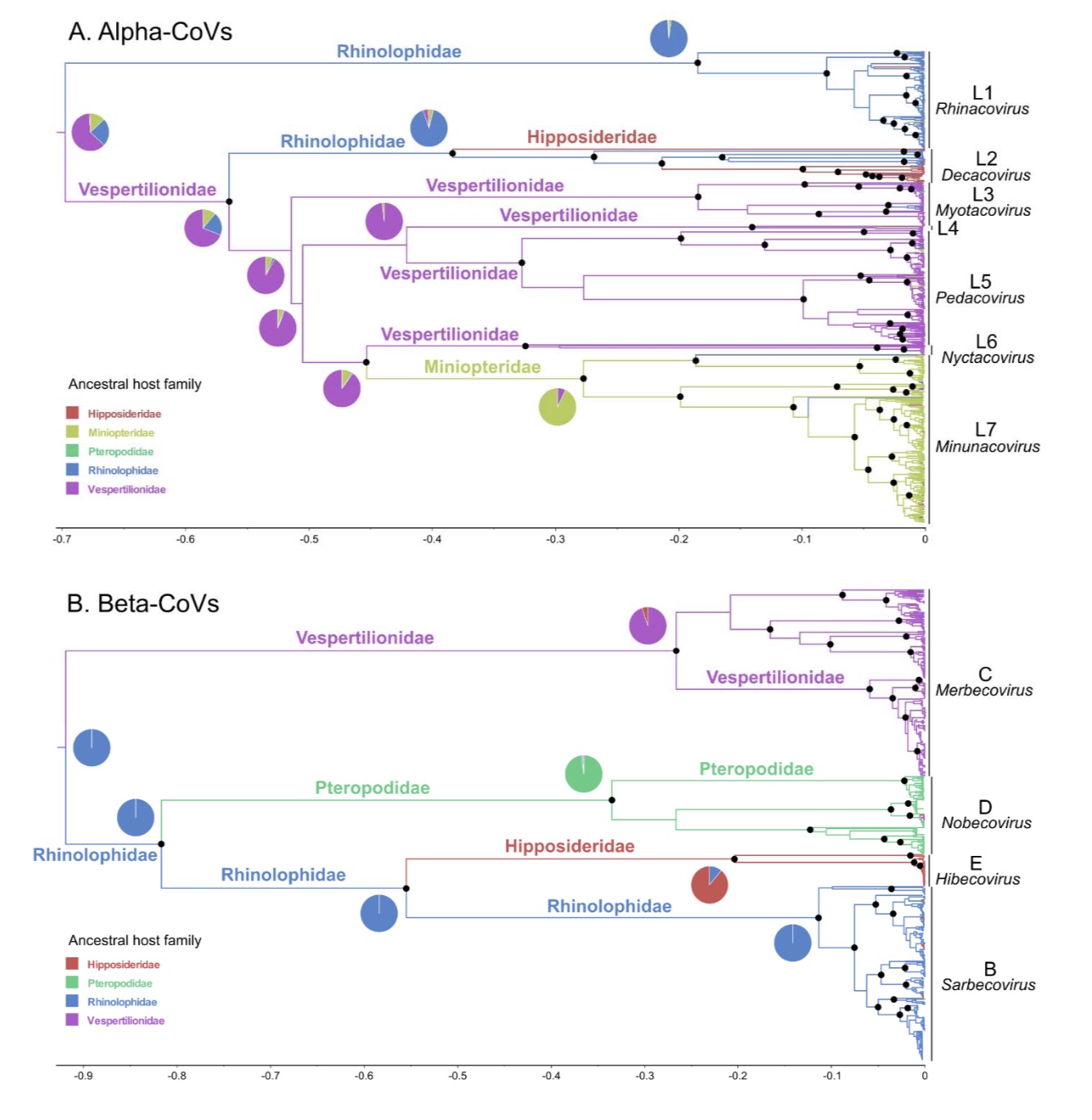
The phylogenetic tree of alpha-coronavirus shows that bat alpha-coronavirus is composed of seven families (L1-L7), corresponding to different subgenus. The first α-coronavirus family corresponds to the Rhinacovirus subgenus, which originates from rhinologid bats. It contains the coronaviruses related to the bat coronavirus HKU2 and SADS.
Another 6 pedigrees originated from vespertilionid bats. The subgenus Decacovirus mainly includes the coronaviruses associated with the HKU10 from the genus Pachycephadidae and the ungulate family. In addition, Myotocovirus, Pedacovirus and another subgenus that has not been confirmed include coronaviruses related to HKU6, HKU-10, and 512 from the bat family. The Nyctacovirus subgenus includes coronaviruses from the bat family, and the Minunacovirus subgenus includes coronaviruses such as HKU7, HKU8, 1A, and 1B from the long-winged bat family.
The research team believes that these 7 families show that frequent cross-species transmission events occur between bats, and the Jutidae and bats are most likely to be bat alpha-crown The ancestral host of the virus.
Beta-Coronavirus is composed of four subgenus. The subgenus Merbecovirus uses the bat family as a host, including the MERS-associated coronavirus, HKU4, and HKU5 coronavirus; the subgenus Nobecovirus comes from the genus Pteropodidae, including the HKU9 coronavirus; the subgenus Hibecovirus comes from the family Hymenoptera; the subgenus Sarbecovirus mainly includes the genus Aster SARS-associated coronavirus and HKU3 coronavirus in bats.
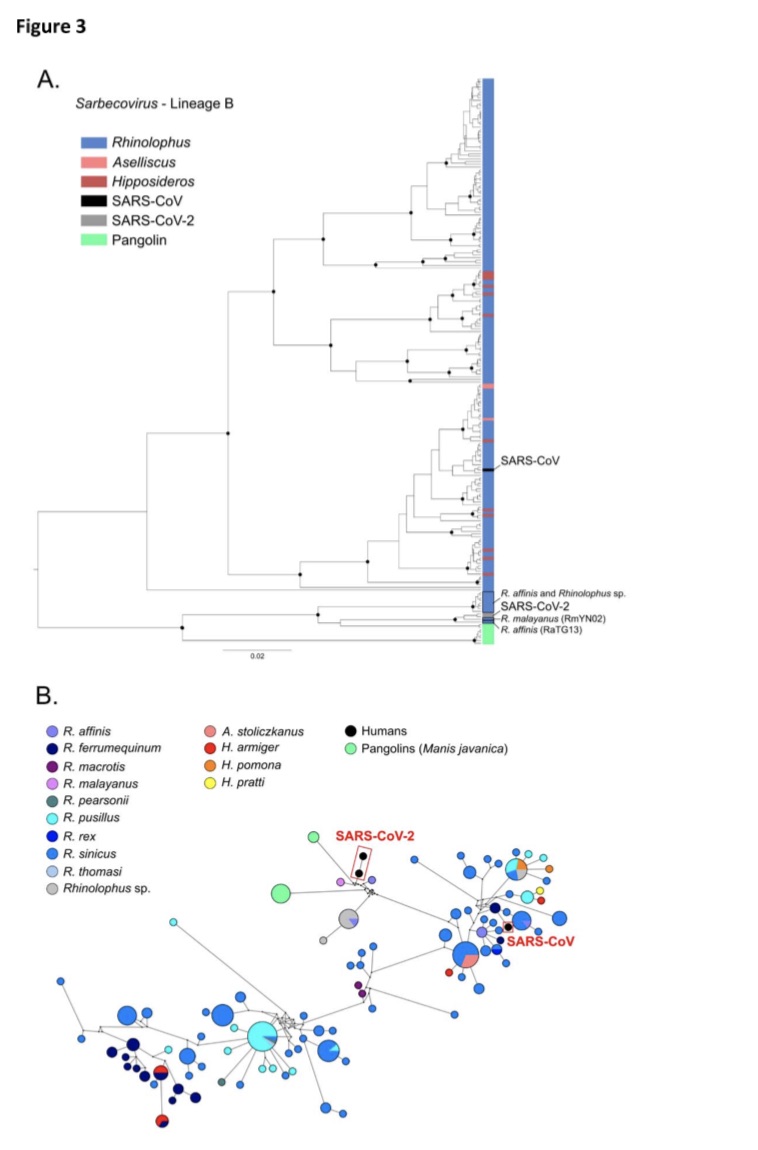
The research team also pointed out that in the evolutionary tree of the Sarbecovirus subgenus, the new coronavirus SARS-CoV-2 and the coronaviruses from the Malayan-headed bat, the middle-headed bat and the pangolin Kinship is recent.
The research team believes that the evolutionary ancestral host of bat beta-coronavirus may also be the genus Pachycephalus and Batidae.
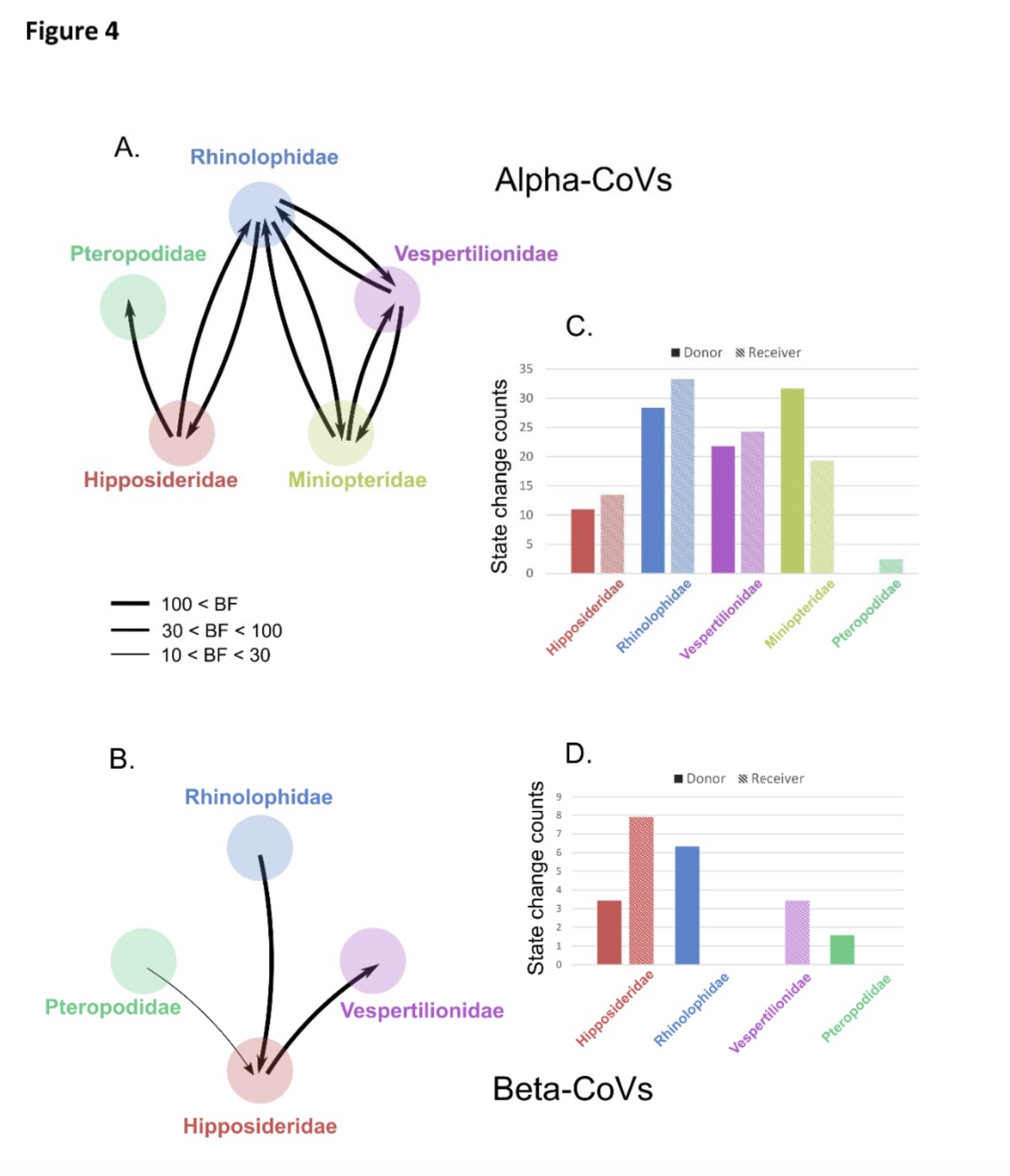
The research team also compared the cross-species transmission of alpha-coronavirus and beta-coronavirus. The results showed that the incidence of cross-family transmission of α-coronavirus was five times that of β-coronavirus. Among α-coronaviruses, cross-family transmissions from Compositae and Longwing bats are more common; while for β-coronaviruses, cross-family transmission from Compositae is most common.
The research team also used the Bayesian discrete geographic model to study the spread of Chinese bat coronavirus in different regions. They divided China into 6 animal geographic regions: Southwest (SW) , Northern Region (NO), Central and Northern Region (CN), Central Region (CE), Southern Region (SO) and Hainan Island (HI).
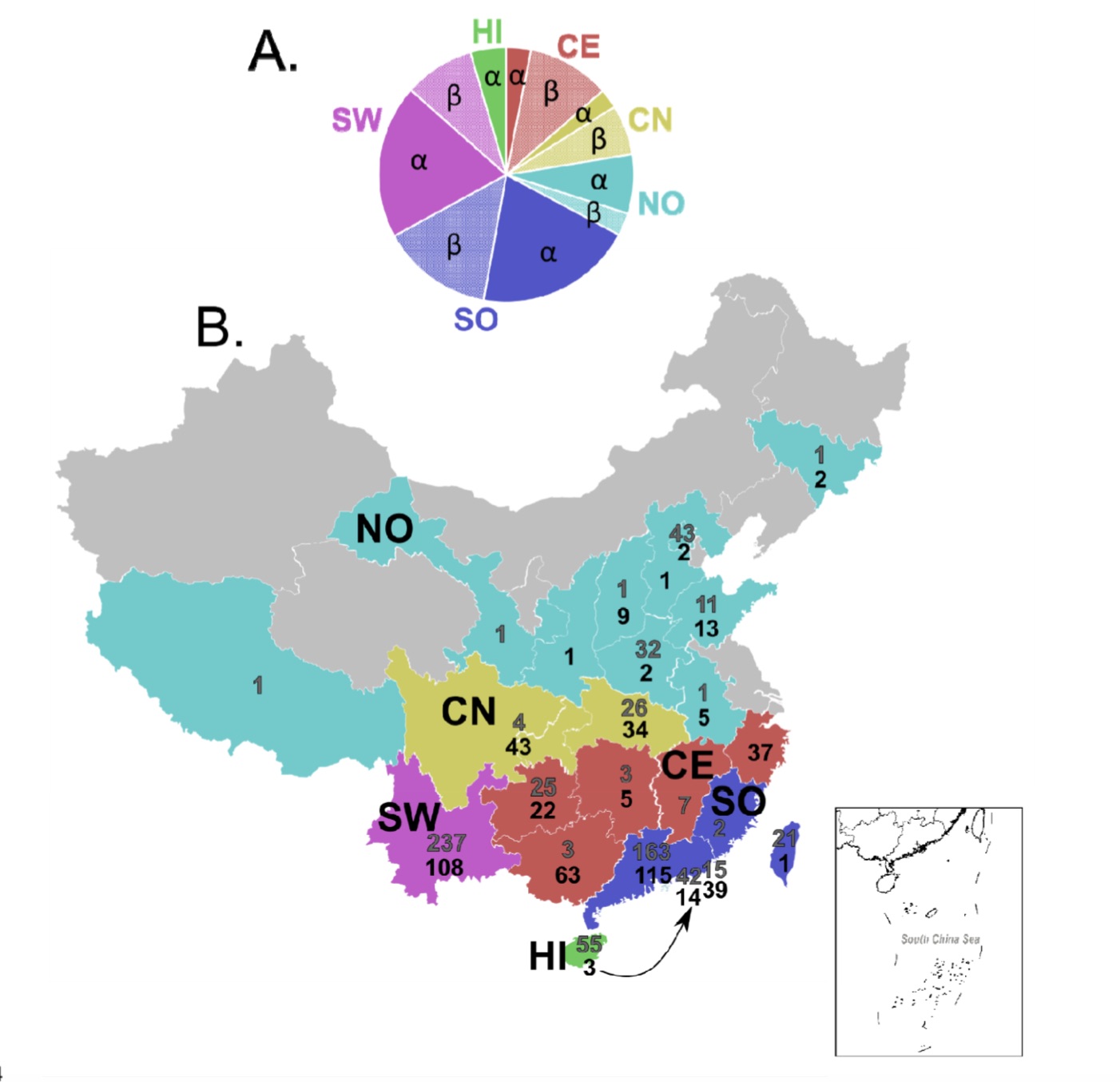
The results show that among α-coronaviruses, members of the Rhinacovirus subgenus including HKU2 and SADS coronaviruses may have originated in the southern region; other α-crownsRhabdovirus may originate in the southwestern region and spread to other regions.
In terms of beta-coronavirus, the southern region may be Merbecovirus subgenus (including HKU4, HKU5 coronavirus) and Sarbecovirus subgenus (including HKU3 and SARS related coronavirus) Place of origin. Nobecovirus (HKU9) and Hibecovirus subgenus may originate in the southwest region. The β-coronavirus may subsequently spread from the southern and central regions to other regions; recently it has also been observed to spread from the northern region to the south.
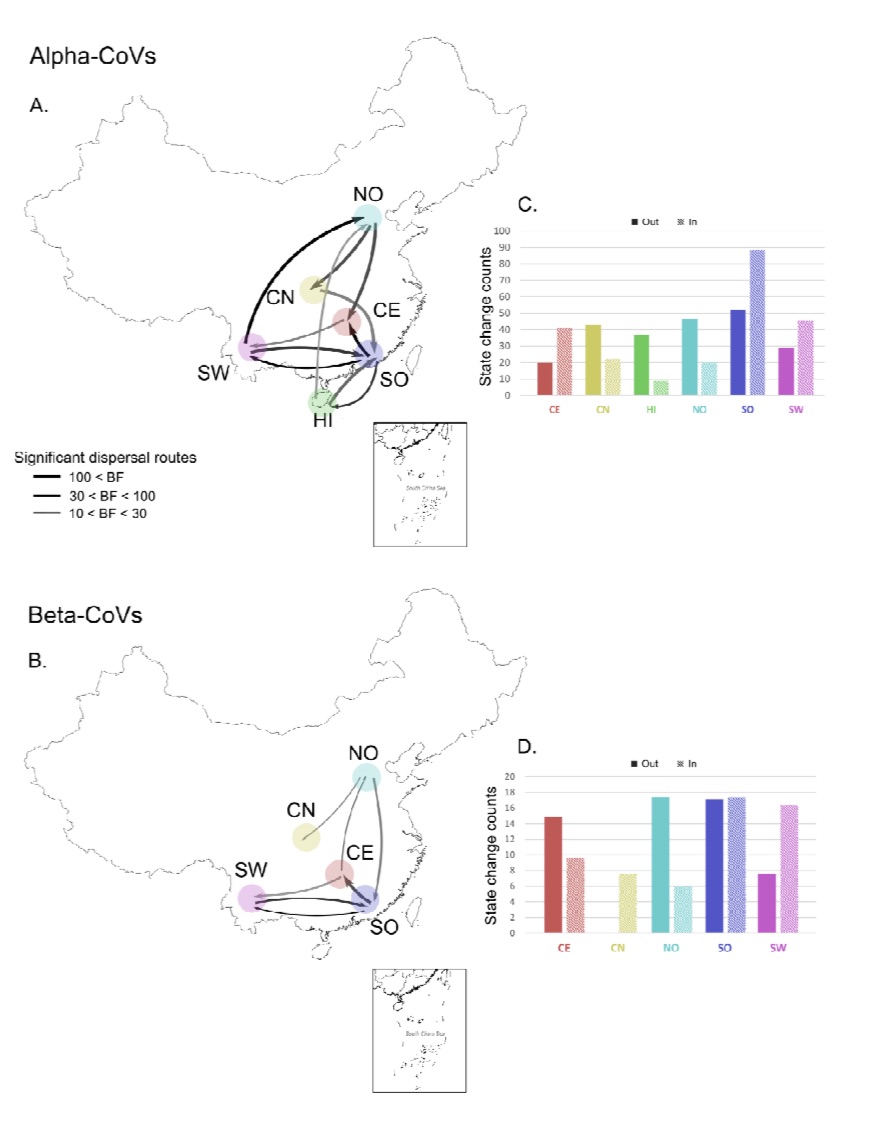
The research team estimated the migration rate per unit of time. The alpha-coronavirus is more than twice the beta-coronavirus, and the southern region is the alpha-coronavirus is the beta-coronavirus total migration event The most frequented area. Alpha-coronaviruses have the most emigration and emigration events in the southern region, beta-coronaviruses have the most emigration events in the northern and southern regions, and the most emigration events in the southern and southwest regions.
In general, the research team believes that the chrysanthemum bat plays an important role in the historical evolution and cross-species transmission of Chinese bat alpha-coronavirus.
In addition, they emphasized that for domestic, future sampling and virus discovery should focus on the two hot spots of coronavirus diversification in southern and southwestern China, Also pay attention to neighboring countries that live like bat species. They mentioned that Myanmar, Laos, Vietnam or other Southeast Asian countries cannot be excluded as the origin of the SARS-CoV-2 ancestral virus.